Crónica Neuronal: matrices de expresión genética para leucemia
Bienvenidos a un nuevo episodio de la serie Crónica Neuronal. Hoy vamos a tocar un problema del campo de la bioinformática. En concreto, vamos a usar matrices de expresión genética para identificar si un paciente de leucemia la tiene de tipo ALL o de tipo AML. Ambas leucemias tienen síntomas muy parecidos y es interesante poder encontrar un modelo de aprendizaje automático que pueda distinguirlas.
¿Qué es una matriz de expresión genética?
La expresión genética es el grado en que un gen se manifiesta en la formación de una proteína, que luego tiene efectos en el organismo. Este grado se mide a través de la presencia de mRNA, aunque no es exactamente proporcional, suele ser adecuado en muchas ocasiones.
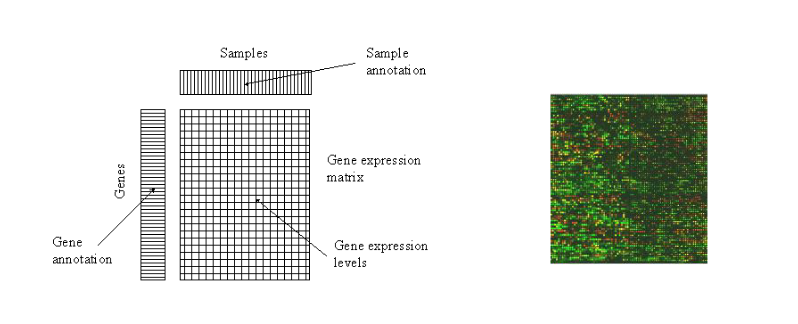
Una matriz de expresión genética no es más que un conjunto de muestras (samples), donde se analiza la expresión genética de muchos genes. Cada casilla representa la expresión genética de un gen en concreto en un individuo en particular. La idea es, con esta información, obtener ciertos patrones de genes que nos ayuden y nos den pistas en la investigación de enfermedades. También nos puede servir para diagnosticar a individuos nuevos enfermedades, así como su estado y poder así obtener mejores tratamientos.
En general, este tipo de datos son complejos de analizar por aprendizaje automático, ya que por lo general existen muy pocos individuos y muchos genes a tener en cuenta. Hay una historia, no demostrada todavía, relacionada con las farmacéuticas que son quienes generan estos ficheros, dice que estas no dan todos los datos que tienen para esforzar a los investigadores a trabajar y luego cuando publican resultados, estas farmacéuticas los pueden comprobar con mucha más facilidad. Un lugar de donde se pueden conseguir matrices de expresión genético es el archivo ArrayExpress mantenido por el European Bioinformatics Institute.
Vistazo a los datos
Para esta crónica neuronal, voy a usar Weka, el software de aprendizaje automático de la Universidad de Waikato (Nueva Zelanda). Es un programa hecho en Java que funciona en Windows, MacOS y Linux.
En este caso, el fichero de datos del que parto ya está en formato ARFF (el nativo de Weka, es similar a CSV pero hipervitaminado) y normalizados (escalado). Una inspección rápida nos hace ver que hay 7129 atributos diferentes, sin nombre, solo están numerados (son los genes). Hay una clase, de tipo binario: ALL y AML. Es decir, para cada paciente de leucemia se nos da su expresión genética de 7129 genes y se nos da su tipo de leucemia real. Solo tenemos 72 pacientes para entrenar.
Vamos a usar validación cruzada con 10 pliegues para el test de los modelos. Vamos a probar los siguientes algoritmos: J48, NaiveBayes, IBK1, Regresión Logística, Perceptrón Multicapa con una capa oculta de 10 neuronas y SVM con kernel lineal.
Si no has usado Weka antes, los pasos para realizar esto son realmente simples. Dirígete a la pestaña "Classify". Una vez allí clica sobre "Choose" y elige el algoritmo de entre las diferentes carpetas.
Si el algoritmo tuviese parámetros de ajuste, se haría clic sobre el nombre del algoritmo en negrita una vez seleccionado.
Pero en la gran mayoría de los algoritmos usaremos los ajustes por defecto. Los resultados son los siguientes:
| Algoritmo | Tasa de Acierto |
| J48 | 0.7916 |
| NaiveBayes | 0.9861 |
| IBK1 | 0.8472 |
| Regresión Logística | 0.9027 |
| MLP (H10) | 0.9722 |
| SVM (lineal) | 0.9861 |
Los mejores resultados los obtienen SVM y NaiveBayes. Esto último nos puede decir que no hay muchos genes correlacionados, sino que son independientes entre sí. Regresión Logística y MLP han tardado considerablemente más que el resto.
Ahora vamos a ver qué atributos son más importantes. Es lógico pensar que muchos de estos atributos (genes) son superfluos, y no afectan en el diagnóstico del tipo de leucemia.
A continuación vamos a analizar qué genes son más relevantes para la tarea de construir un clasificador entre los tipos de leucemia.
Para ello primero usaremos métodos de filtrado.
Filtrado
Los métodos de filtrado se basan en diferentes heurísticas que nos permiten seleccionar un conjunto de atributos relevantes. La mayoría de estas heurísticas elaboran un ránking, dentro de las cuáles podemos elegir el número de atributos que queramos. Los algoritmos para el filtrado que vamos a probar son: Incertidumbre simétrica, ReliefF, SVM recursivo y CfsSubset (este último no genera ranking).
|
Algoritmo |
Atributos (4) |
|
Incertidumbre simétrica |
0.74 1834 attribute1834 0.74 4847 attribute4847 0.737 1882 attribute1882 0.734 3252 attribute3252 |
|
ReliefF |
0.26503581944 3252 attribute3252 0.25909453333 4196 attribute4196 0.21482608611 1779 attribute1779 0.19528160278 4847 attribute4847 |
|
SVM |
7129 1882 attribute1882 7128 1834 attribute1834 7127 1779 attribute1779 7126 1796 attribute1796 |
|
CfsSubset |
No se pudo calcular |
Si nos quedamos solo con cuatro atributos, los resultados son los siguientes. Estos resultados se obtienen en Weka en la pestaña de Selección de Atributos. Eligiendo el algoritmo y un método de búsqueda (Ranker, o en su defecto, GreedyStepwise).
Podemos repetir el procedimiento con 8, 16 y 32 atributos. No os voy a poner los atributos, pero sí vamos a ver como se comportan.
Vemos que en general hay genes que se repiten entre métodos, como el 1779 o el 4847. Estos genes pueden ser determinantes para diagnosticar los diferentes tipos de leucemia.
|
|
J48 |
NaiveBayes |
IBK1 |
Reg. Log |
MLP (H10) |
SVM (lineal) |
|
Incertidumbre simétrica (4) |
0.9027 |
0.9444 |
0.9166 |
0.9444 |
0.9305 |
0.9305 |
|
Incertidumbre simétrica (8) |
0.8472 |
0.9444 |
0.9305 |
0.9444 |
0.9583 |
0.9305 |
|
Incertidumbre simétrica (16) |
0.8472 |
0.9583 |
0.9583 |
0.9583 |
0.9861 |
0.9444 |
|
Incertidumbre simétrica (32) |
0.8611 |
0.9583 |
0.9583 |
0.9583 |
0.9722 |
0.9722 |
Con solo 4 atributos seleccionados por incertidumbre simétrica, obtenemos muy buenos resultados con algunos métodos: NaiveBayes y Regresión Logística.
|
|
J48 |
NaiveBayes |
IBK1 |
Reg. Log |
MLP (H10) |
SVM (lineal) |
|
ReliefF (4) |
0.9166 |
0.9166 |
0.8888 |
0.9444 |
0.9444 |
0.9444 |
|
ReliefF (8) |
0.8611 |
0.9722 |
0.9444 |
0.9027 |
0.9305 |
0.9444 |
|
ReliefF (16) |
0.8472 |
0.9444 |
0.9305 |
0.9305 |
0.9305 |
0.9722 |
|
ReliefF (32) |
0.8333 |
0.9583 |
0.9305 |
0.9583 |
0.9722 |
0.9722 |
En este caso con 4 y 32 atributos se obtienen resultados similares al método anterior. No obstante, los algoritmos con buen desempeño son diferentes.
|
|
J48 |
NaiveBayes |
IBK1 |
Reg. Log. |
MLP (H10) |
SVM (lineal) |
|
SVM (4) |
0.9166 |
0.9722 |
1 |
1 |
1 |
1 |
|
SVM (8) |
0.9166 |
0.9722 |
0.9861 |
0.9861 |
1 |
1 |
|
SVM (16) |
0.875 |
1 |
1 |
1 |
1 |
1 |
|
SVM (32) |
0.8472 |
0.9861 |
1 |
1 |
1 |
1 |
La selección por SVM es excelente, ya que con solo 4 genes, logra tasas de acierto del 100% en 4 métodos diferentes. Seguramente estos 4 genes tengan la relación más directa con la enfermedad.
Envoltorio
Otro método de selección de atributos es el método del envoltorio. Aquí se elige un algoritmo de clasificación y según su desempeño se van descartando atributos irrelevantes. Idealmente, el mismo algoritmo envuelto es el que luego se usa en clasificación, pero aquí probaremos todos con todos.
|
Algoritmo |
Atributos |
|
J48 |
Selected attributes: 4847 : 1 attribute4847 |
|
NaiveBayes |
Selected attributes: 6,461,760,6615 : 4 attribute6 attribute461 attribute760 attribute6615 |
|
IBK1 |
Selected attributes: 28,1834,3258,3549 : 4 attribute28 attribute1834 attribute3258 attribute3549 |
|
Reg. Log. |
Selected attributes: 202,1882,6049 : 3 attribute202 attribute1882 attribute6049 |
|
MLP (H10) |
Selected attributes: 1796,1834,2288 : 3 attribute1796 attribute1834 attribute2288 |
|
SVM (lineal) |
Selected attributes: 162,1796,2111,3252 : 4 attribute162 attribute1796 attribute2111 attribute3252 |
En general la mayoría de envoltorios se decantan por 3 o 4 atributos, pero muchas veces diferentes. J48 se decanta por solo un único atributo.
|
|
J48 |
NaiveBayes |
IBK1 |
Reg. Log. |
MLP (H10) |
SVM (lineal) |
|
WrapJ48 |
0.9444 |
0.9305 |
0.9166 |
0.9305 |
0.9444 |
0.8611 |
|
WrapNB |
0.9166 |
0.9861 |
0.8333 |
0.9583 |
0.9583 |
0.8194 |
|
WrapIBK1 |
0.8888 |
0.9861 |
1 |
0.9444 |
0.9444 |
0.8888 |
|
WrapRegLog |
0.9583 |
0.9861 |
0.9444 |
1 |
0.9722 |
0.8333 |
|
WrapSVM |
0.8888 |
0.9305 |
0.9166 |
0.9444 |
0.9861 |
0.9722 |
En general el comportamiento es bueno con el método que hizo de envoltorio, y peor en el resto. Sorprende MLP (H10) que logra resultados muy decentes con todas las selecciones y SVM, que da resultados muy mediocres salvo con su envoltorio.
Si comparamos métodos de filtro con métodos de envoltorio, nos encontramos con que hemos visto métodos de filtro muy superiores como SVM, si bien el método de envoltorio con el mismo algoritmo en ambas etapas es también una opción muy interesante.
Por norma general, los métodos de envoltorio son mejores pero también mucho más costosos de ejecutar. Es por ello, que ante las selecciones de filtrado obtenidas antes, normalmente nos quedaríamos con esos atributos. En este caso, y por mera curiosidad, hemos continuado con los métodos de envoltorio, mucho más lentos.
Si tuviésemos que construir un sistema hoy para detectar entre los tipos de leucemia, la opción sería eliminar atributos mediante SVM recursivo y construir el clasificador bien con SVM o con otro de los algoritmos que aciertan siempre, ya que no tenemos otros criterios todavía para discernir.
Espero que os haya gustado este episodio de Crónica Neuronal. En este caso, no he usado Python sino que he usado Weka, pero espero que aporte perspectiva y ayude a la gente a conocer por un lado los arrays de expresión genética, y por otro, los algoritmos de selección de atributos relevantes, sin entrar en el detalle de como funcionan.